Gentechnik-Projekt im Labor
1.) Sicherheitsvorkehrungen
gefährliche Chemikalien
·
Ethidiumbromid: lagert sich zwischen der DNA ein. Bei der Bestrahlung mit UV-Licht wird es sichtbar und wird deshalb zur Färbung der DNA verwendet.
·
SDS: unterstützt das Aufbrechen der Zellen.
·
Guanidiniumthiocyanat: wird zum Aufschließen der Zelle verwendet, d.h. es bricht die Zellwände auf.
·
CTAB: wie oben.
·
Chloroform: ist ein organisches Lösungsmittel, und es werden damit die Proteine (z.B. Histone) von der
DNA getrennt.
Da diese Chemikalien gesundheitsschädlich sind, ist es notwendig, Handschuhe zu tragen und unter dem Abzug zu
arbeiten.
2.) Analyse von Gensoja
Am ersten Tag im Labor untersuchten wir Sojamehl. Dabei hatten wir
3 Proben von der Firma Fluka. (Sojamehl mit 0%, 0,1% und 2% gentechnisch verändertem Material) zur Verfügung.
Zur Unterscheidung von gentechnisch verändertem Soja von
"normalen" Soja suchen wir einen Abschnitt des DNA Stranges, der
der Pflanze bei der Genmanipulation eingebaut wurde. Der Abschnitt, nach dem Promotor und der Signalsequenz, ist bei einer natürlichen
Pflanze nicht vorhanden.
|

|
Aufbrechen der Zellen:
Um an die gewünschte DNA zu gelangen, müssen zuerst die Zellen
mit Hilfe eines Extraktionspuffers aufgeschlossen werden.
b) Reinigen der DNA:
- Reinigen der DNA von Zellwandresten durch Zentrifugieren
- Reinigen der DNA von Proteinen mit Hilfe von Chloroform ( Trichlormethan).
Dieses organische Lösungsmittel denaturiert die Proteine ( Zerstörung der Tertiärstruktur) und somit sind sie nicht mehr löslich und fallen aus.
- Die DNA wird, durch Zugabe von 100% Isopropanol und danach durch 70 % Ethanol, konzentriert und von Salzen gereinigt.
c) PCR (Polymerase-Ketten-Reaktion):
|
Da für die folgenden Arbeitsvorgänge zu wenig DNA vorhanden ist, muß diese
vermehrt werden. Dies geschieht in der PCR-Maschine (Firma Techne Modell PHC-2).
Bevor man diesen Vorgang direkt an der Maschine starten kann, müssen einige
Vorbereitungen getroffen werden.
7 DNA-Ansätze wurden für die Analyse (Stock solution) in Eppendorfs (E) gefüllt.
In alle wurden Primermengen hineinpipettiert. Der Primer ist das Steuerelement, das
den Startpunkt der DNA-Verdopplung festlegt.
|
| |
Primer 1 (p35s-f2) |
Primer 2 (petu-r1) |
GACT (je 5 µl von 2 mM) 1:10 |
10 x Puffer |
Polymerase
2U/µl |
A. bidest. |
Summe |
|
Pro Eppendorf |
0,2 µl |
0,2 µl |
20 µl |
5µl |
0,43 µl |
22,6 µl |
48,43µl |
|
Ges.-Vol.: |
1,4 µl |
1,4 µl |
140 µl |
35 µl |
3 µl |
158,5 µl |
339,25 µl |
Proben in Gesamtvolumen pro Eppendorf von 50µl:
|
1 | 0% | 1µl
|
|
2 | 0% | 3µl
|  |
|
3 | 0,1% | 1µl
|
|
4 | 0,1% | 3µl
|
|
5 | 2% | 1µl
|
|
6 | 2% | 3µl
|
|
7 | Leerprobe (keine DNA)
|
Die Maschine arbeitet in drei Verfahrensschritten, wobei sie "lediglich" die Temperaturen regelt:
- PCR-Maschine erhitzt auf 93°C. Dadurch wird der Doppelstrang der DNA in zwei Einzelstränge
gespalten, weil die Bindung zwischen den Basen keinen hohen Temperaturen standhält.
- Danach kühlt die Maschine auf 55°C ab. Bei dieser Temperatur lagern sich die
Primer (Ansatzstelle der Polymerase) an.
- Jetzt wird wieder auf 72°C erhitzt, damit die Polymerase ein bestimmtes DNA - Stück vervielfacht.
- Nun beginnt der Zyklus von vorne: Die Probe wird wenige Sekunden auf 92°C erhitzt, damit die
gebildeten Doppelstränge wieder getrennt werden usw. ...
Dieser Vorgang wird in unserem Falle 40 x wiederholt, der insgesamt ca. 3,5 Stunden dauert.
d) Gießen des Agarosegels:
Während die PCR-Maschine arbeitet, wird das Gel für die Elektrophorese vorbereitet.
Dazu muß man den Schlitten, in den dann später das Gel gegossen wird, am oberen und unteren Rand mit einem
Klebeband zukleben, um das Herausfließen des flüssigen Gels zu verhindern. Danach steckt man in die vorgesehenen
Schlitze am Schlitten die Kämme hinein. Dann beginnt man mit der Herstellung des Gels.
Zuerst gibt man 2g von dem Agarosepulver in einen Erlenmeyer–Kolben und füllt es mit einer 1x TAE Lösung auf.
Dann wird dieses Gemisch bis zum Kochen erhitzt.
Gleichzeitig löst sich das Agarosepulver in der TAE
Lösung. Hat sich das Pulver vollständig gelöst, kühlt man
die Lösung leicht ab. Um nachher die Balken der DNA
unter UV-Bestrahlung erkennen zu können, wird
Ethidiumbromid dazugegeben. Nun muß die Lösung mit
dem Ethidiumbromid geschüttelt werden, daß das
Ethidiumbromid gleichmäßig verteilt ist. Da das ganze
Gemisch noch warm (flüssig) ist, kann man es zügig in den
vorbereiteten Schlitten gießen. Mit der Pipette versucht man dann entstandene Luftbläschen im Gel zu entfernen.
|

|
Dann läßt man das Gel im Schlitten abkühlen (verfestigen) .
|
Nach Verfestigung des Gels muß das Klebeband
entfernt und der Schlitten in die Pufferlösung der
Elektrophorese eingesetzt werden.
Danach den Kamm vorsichtig senkrecht
herausziehen. Bei Bedarf so viel TAE-Puffer
nachgießen, daß das Gel vollständig bedeckt ist.
Nun können die vorbereiteten Probelösungen, die
Marker (DNA-Stückchen mit definierter Länge)
und Proben von genomischer Soja-DNA in die Slots
einpipettiert werden. Bei einer Spannung von 80
|
Volt wird die Elektrophorese 1,5 Stunden gefahren.
|
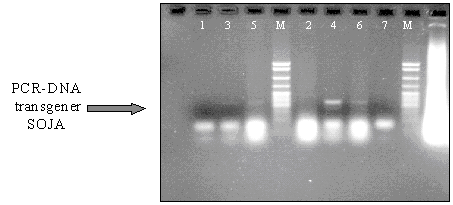
Ergebnis: Die gesuchte Bande
war bei allen Gensojaprodukten
vorhanden, manche deutlich
sichtbar, manche nur schwach.
Der Nachweis wurde also
gebracht.
|
|
3). Hasenkotanalyse
1. Zellaufschluß
Der Kaninchenkot, Meerschweinchenkot, Spinat und Salat werden in flüssigem Stickstoff, bei -196°C gefroren.
Danach wird der Kot und das Gemüse mechanisch zerkleinert. Dadurch entsteht feiner Staub. Dem Pulver wird
Exktraktionspuffer beigemengt. Dieser ist hochsalzig und durchlöchert die Zellwände.
2. Reinigung der DNA
Diese Probe wird nun zehn Minuten gekocht. In zwei Eppendorfgefäße kommen je 400 m
von der aufgekochten
Lösung. Darauf werden die festen Bestandteile durch Zentrifugieren vom flüssigen Teil getrennt. Vom Überstand

|
werden 250 µl von den Lösungen mit jeweils 50 µl
Pufferlösung vermischt. Durch Vermengung der
Siliziumkügelchen mit der Lösung bildet sich eine milchige
Flüssigkeit. Die DNA bindet sich an die Siliziumkügelchen
(Glasperlen). Damit sich die DNA gleichmäßig bindet,
werden die Proben geschüttelt ("Vortexen"). 10 min müssen
die Proben bei Raumtemperatur stehen gelassen, und
schließlich 30 sek. zentrifugiert werden. Den Überstand
schüttet man rasch in den organischen Abfall. Die an den
Silizium-Kugeln gebundene DNA wird jetzt gewaschen:
Dazu gibt man 300µl Waschpulver. Daraufhin werden die
Proben 5 sek. geschüttelt, 30 sek. zentrifugiert und der
entstehende Überstand (Zellreste, Proteine) abgeleert.
Dieser Vorgang wird zweimal durchgeführt.
Die DNA bindet sich nur an die Glaskugeln, weil eine sehr
hoch salzige Umgebung in der Probe vorherrscht. Nachdem
die Lösung gereinigt worden ist, gibt man 70%igen kalten
Ethanol dazu (-20°C). Der Überstand, indem sich noch
restliche Salze befinden, wird wiederum abgeschüttet.
Danach wird das Pellet im Exsikkator getrocknet. Die
Zugabe von TE-Puffer bei 70°C bewirkt, daß sich die
Glasperlen von der DNA trennen, da der Puffer eine
|
niedrige Salzkonzentration hat. Die Probe wird 15 min in 56°C- Wärmeblock gestellt. Nun wird wieder zentrifugiert
und die Glasperlen setzen sich am Boden ab. Oben schwimmt die reine DNA. Diese wird wieder in neue Eppendorfs
hineinpipettiert.
|
10 x Puffer |
GACT (je 5µl von 2 mM) |
Primer 1
1:20 |
Primer 2
1:20 |
BSA
1 mg /ml |
Polymerase Dynazyme und Vent |
Aqua Bidest |
Summe |
|
Je Eppendorf |
5 µl |
20 µl |
2 µl |
2 µl |
0,1 µl |
0,5 µl |
19,5 µl |
49,1 µl |
|
Gesamtvolumen |
45 µl |
180 µl |
18 µl |
18 µl |
0,9 µl |
4,5 µl |
175,5 µl |
|
Proben:
|
1 |
Spinat |
1:10 |
1µl |
 |
|
2 |
Spinat |
1:20 |
1µl |
|
3 |
Kot (mit Spinat) |
1:10 |
1µl |
|
4 |
Kot (mit Spinat) |
1:20 |
1µl |
|
5 |
Salat |
1:10 |
1µl |
|
6 |
Salat |
1:100 |
1µl |
|
7 |
Kot (mit Salat) |
1:10 |
1µl |
|
8 |
Kot (mit Salat) |
1:20 |
1µl |
|
9 |
Leerprobe (keine DNA zugefügt) |
|
|
Das PCR- Produkt wird nun mit Hilfe von Agarose- Gelelektrophorese
aufgetrennt (1,2% Agarose/ 90 V/ 30min). Dieser Vorgang zeigt, ob und wieviel DNA vorhanden ist.
Durch dieses Verfahren wird sichtbar, welche
Probe positiv (DNA vorhanden) und welche
negativ ist. Um die Menge der DNA zu erhöhen,
werden die positiven, gleichwertigen Lösungen
vermengt.
Die Probe des Meerschweinchen- Kotes (Kotsalat)
war negativ.
Auch beim Hasenkot (Kotspinat) waren , im
Gegensatz zu der Spinat- und Salatproben nur
geringe Anteile der DNA vorhanden.
Das Ergebnis können wir im Bezug auf die reinen
|
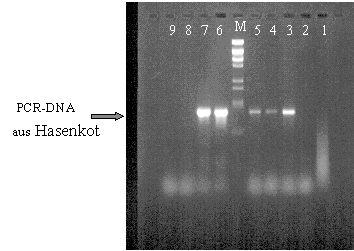
|
Spinat und Salatproben dadurch begründen, daß in reinen Pflanzenteilen mehr DNA vorhanden ist, als in verdauten
Substanzen.
Restriktionskartierung (Verdau mit Restriktionsenzymen)
Für diesen Vorgang brauchen wir reine DNA:
Zuerst werden die Proben mit 2fach destilliertem Wasser (A.bidest) auf gleiche Mengen gebracht (360 m
l).
Die Reinigung der PCR- Probe:
- Das Gemisch von DNA, NaCl und 100%ige Ethanol wird 20 min auf Eis gestellt. Dadurch fällt die DNA aus.
- Danach werden die Proben 15 min bei 14000 Umdrehungen/min zentrifugiert. Dadurch setzt sich die gefällte
DNA am Boden ab und der Überstand kann abgeleert werden.
- Um die Salze von der DNA zu trennen gibt man 70%ige EtOH hinzu. Die Probe wird kurz geschwenkt,
zentrifugiert und der Überstand abgeleert.
- Nun wird die DNA unter Vakuum in einem Exsikkator getrocknet und danach in 30 m
l A. bidest gelöst.Vor der Verdauung untersucht man, mit Hilfe der Gelelektrophorese, ob sich noch DNA in der Probe befindet.
Nun beginnt die eigentliche Restriktionskartierung.
Dazu werden verschiedene Enzyme und dazu passende Puffer benötigt. Jedes Enzym schneidet nach einer bestimmten Basenabfolge auf der DNA.
Da der Kaninchenkot (Kot-Spinat) zu wenig DNA enthielt, verwendeten wir anstatt fünf nur zwei Enzyme, damit für diese Enzyme genug DNA vorhanden ist. Dann werden die Salatproben und Spinatproben auf jeweils fünf Eppendorf aufgeteilt. Dazu kommmt in jedes E. 2µl eines Enzyms und 2µl des dazugehörigen Puffers. Die Menge der Probelösung und des Wassers auf Grund der Menge der DNA kommt.
Restriktionsenzyme:
|
Enzyme |
Puffer |
Temperatur |
|
Hae 3
|
M |
37° C
|
|
Mae 3
|
Mae 3 Spezial |
55° C
|
|
Sac 2
|
4 |
37° C
|
|
Rsa 1
|
1 |
37° C
|
|
Cfo 1
|
L |
37° C
|
Daraus ergibt sich folgende Tabelle:
|
N° |
Probe
|
Menge der
Probelösung |
A. bidest |
Enzym |
Puffer |
|
1 |
Hae 3 Salat |
4 m
l |
12 m
l |
2 m
l |
2 m
l |
|
2 |
Mae 3 Salat |
4 m
l |
12 m
l |
2 m
l |
2 m
l |
|
3 |
Sac 2 Salat |
4 m
l |
12 m
l |
2 m
l |
2 m
l |
|
4 |
Rsa 1 Salat |
4 m
l |
12 m
l |
2 m
l |
2 m
l |
|
5 |
Cfo 1 Salat |
4 m
l |
12 m
l |
2 m
l |
2 m
l |
|
6 |
Hae 3 Spinat |
4 m
l |
12 m
l |
2 m
l |
2 m
l |
|
7 |
Mae 3 Spinat |
4 m
l |
12 m
l |
2 m
l |
2 m
l |
|
8 |
Sac 2 Spinat |
4 m
l |
12 m
l |
2 m
l |
2 m
l |
|
9 |
Rsa 1 Spinat |
4 m
l |
12 m
l |
2 m
l |
2 m
l |
|
10 |
Cfo 1 Spinat |
4 m
l |
12 m
l |
2 m
l |
2 m
l |
|
11 |
Hae 3 Kot- Sp. |
13,5 m
l |
2,5 m
l |
2 m
l |
2 m
l |
|
12 |
Cfo 1 Kot-Sp. |
13,5 m
l |
2,5 m
l |
2 m
l |
2 m
l |
Nachdem die Substanzen jeweils in ein Eppendorf pippetiert worden sind, werden sie zentrifugiert. Daraufhin werden sie eine Stunde auf der für die Enzyme nötige Temperatur gebracht und gehalten. Danach ist der Restriktionsenzymvorgang (Schneiden der DNA) abgeschlossen. Um nun sicher zu gehen, daß kein Enzym weitere DNA schneidet, wird ein Hemmstoff (EDTA) hinzugefügt. Dieses EDTA bewirkt, daß zweiwertige Ionen abgefangen werden können, die für die Enzyme lebensnotwendig sind.
Da der Arbeitstag zu Ende ging, mußten wir die DNA auf –20°C legen, um das Überleben der DNA zu sichern.
Am nächsten Tag tauten wir die Proben auf, versahen sie mit 2 m
l Laufpuffer und zentrifugierten kurz die Gemische.
Als letzten Arbeitsschritt trugen wir die Proben auf das Elektrophoresegel auf.
Nach einer Wartezeit von 2,5 Stunden konnten wir das durchaus positive Ergebnis unter dem UV-Licht betrachten und ablesen.